complete project
MethylNet
Scientific Premise:
The development of and access to DNAm methods and data is becoming more widespread. DNA Methylation (DNAm) deep learning technologies have been shown to accurately characterize tissue and disease states from high-dimensional biomarkers through unsupervised embedding, generation, classification and regression tasks, while traditional DNAm association studies localize and group methylated cytosine-guanine dinucleotides (CpGs) into genomic contexts pertinent to the study and then associate these contexts with the outcome. However, deep learning approaches, which in many cases outperform the traditional statistical approaches, are still largely unable to elucidate how these contexts relate to diagnosis and prognosis. We aspire to develop methods which make DNAm deep learning methods more interpretable and accessible to the epigenetic community.
Motivation:
Our DNA is organized into 23 pairs of chromosomes, though DNA under normal conditions are organized into seemingly discohesive but topologically associated structures called chromatin, comprised of nucleosomes, where DNA are coiled around protein complexes called histones. For transcription to occur, regions of chromatin between the nucleosomes must be open and accessible. DNA methylation (DNAm) refers to the addition of a methyl group to the cytosine basepair in a cytosine guanine dinucleotide, CpGs, which occur at single sites in the genome that sometimes occur in clusters called CpG islands. Alterations that occur in some of these key areas may heavily impact transcription. As an example, alterations to methylation in the promoter island regions are heavily implicated with changes to transcription, where unmethylated CpGs in the promoter island are associated with transcriptional activation of the gene and ethylation to the island is typically associated with transcriptional repression. When study DNAm, we typically study the signal from mixture of cells, where cells within bulk mixture may be methylated or unmethylated at specific sites. We refer to the proportion of methylated alleles at a site as the beta value which is typically close to zero or one to indicate hypomethylation or hypermethylation across the cell mixture accordingly.
DNA methylation may also serve as initiating alteration for oncogenesis (e.g., transcriptional repression of mismatch repair gene, where cancer can arise due to mounting genomic instability ). Thus, we care a lot about identifying DNAm alterations that are associated with phenotypic changes but also usually invariant to cellular composition. The reason why we want changes to be invariant to cellular composition because cell composition for specific tissue may confound disease associations found through epigenome wide association studies (EWAS). DNA methylation can be used to inform cell mixture composition in a bulk specimen through deconvolution algorithms that assume immune cells have characteristic DNAm profiles. Additionally, aging strongly correlated with changes to DNAm, so much so, that it serves as a strong predictor for inferring age when it is unknown. However, the biological age predicted using DNAm does not always agree with our chronological age, potentially indicating accelerated aging. Another interesting fact about DNAm is that it is a reversible somatic alteration, which presents therapeutic implications. If we can develop tools to explain why we age faster biologically, we can develop interventions and therapeutics which can help us turn back time.
We typically measure ~500k at a time using array based technologies. Due to the prohibitive dimensionality, the predictors are highly multi-collinear and interaction space not clearly explored. Most methods consider 1 CpG at time and or used penalized regression procedures, which miss unintuitive relationships and suffer from collinearity issues. Random forest algorithms have been successfully employed but are limited in massive compute time and memory. However, the deep learning models can operate on the order of 1 million CpGs at a time due to availability of graphics processing units. Our lab primarily employs deep learning methods to study DNA methylation.
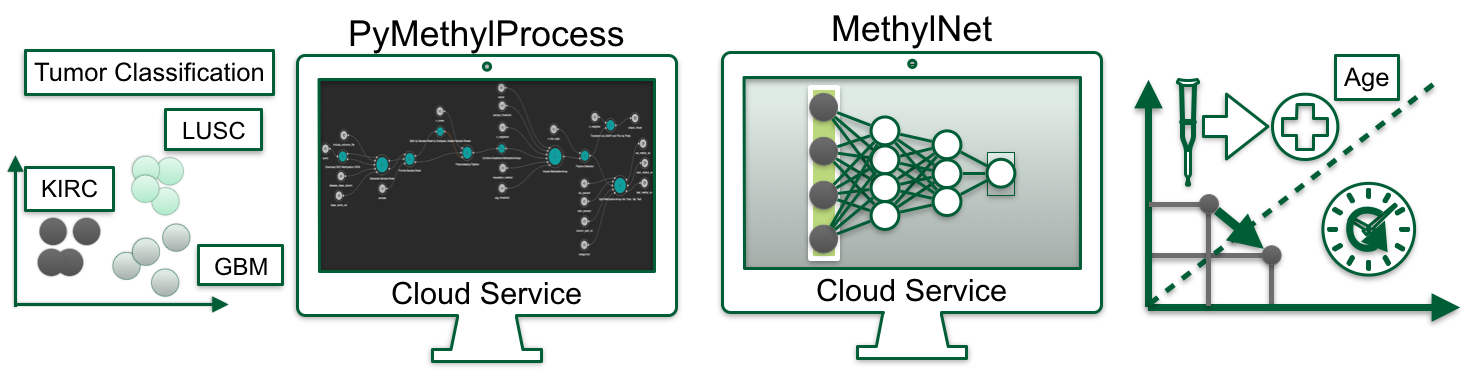
First, we developed workflow automation techniques, PyMethylProcess, which aims to standardize and scale the preprocessing of DNAm data, from signal intensities. to common preprocessing tasks, to generation of low dimensional embeddings using reduction techniques such as UMAP. Concurrently, the platform prepares datatypes that are amenable to machine learning analyses and also utilizes Docker and Common Workflow Language to share reproducible workflows with the greater scientific community. PyMethylProcess has the capacity to preprocess at scale with the click of a button.
Then, we developed a standardized command-line neural network framework, MethylNet, that provide capabilities to uncover meaningful biological features. Particularly, the framework can embed DNAm data (compression from high to low dimensionality), generates new DNAm data through sampling of a posterior latent distribution, and classifies/regresses on DNAm profiles for the prediction of cateogorical/continuous (e.g. inference of disease subtypes, age and cell-type proportion estimation) outcomes. The framework also features a few methods to interrogate the findings to find CpGs correspondent with the outcomes. Finally, the software builds off of previously established deep learning methods for DNAm and interfaces with the data formats introduced by PyMethylProcess. As an example, MethylNet can classify different tumors, estimate proportions of different immune cell types, or estimate an individual’s biological age.
Finally, we developed MethylCapsNet, which is a deep learning framework that builds off of the MethylNet framework to associate CpG-groups with outcomes of interest, much like running a Group LASSO. Finding CpGs that are associated with predicted outcomes of interests
using neural networks can be difficult due to the high dimensional
nature of the data, while sorting CpGs into biologically inspired
groupings as an intermediate neural network layer reduces this
interpretation/validation burden while potentially improving prognostic
performance or at least reducing the complexity/resources required by
the model. As an example of these “capsule” layers, CpGs may map to genes, and gene specific methylation can be related to oncogenesis. Methods I have been exploring incorporate biological structure into the neural network design; for instance, pruning neural network connections such that the signal can only pass from CpGs to genes of interest. These genes are the predictors of interest.
Manuscripts:
- Levy, J. J., Titus, A. J., Salas, L. A., et al. PyMethylProcess—convenient high-throughput preprocessing workflow for DNA methylation data. Bioinformatics 35, 5379–5381 (2019).
- Levy, J. J., Chen, Y., Azizgolshani, N., et al. MethylSPWNet and MethylCapsNet: Biologically Motivated Organization of DNAm Neural Networks, Inspired by Capsule Networks. npj Syst Biol Appl 7, 1–16 (2021).
- Levy, J. J., Titus, A. J., Petersen, C. L., et al. MethylNet: an automated and modular deep learning approach for DNA methylation analysis. BMC Bioinformatics 21, 108 (2020).
- Levy, J. J., Titus, A. J., Petersen, C. L., et al. MethylNet Test Pipeline. (Code Ocean, 2019). doi:10.24433/CO.6373790.V1.